

[ad_1]
Biomolecular dynamics simulations are essential for all times sciences, providing insights into molecular interactions. Whereas classical molecular dynamics (MD) simulations are environment friendly, they lack chemical precision. Strategies like density useful principle (DFT) obtain excessive accuracy however are too computationally intense for giant biomolecules. MD simulations permit commentary of molecular habits, with classical MD utilizing interatomic potentials and ab initio MD (AIMD) deriving forces from digital constructions. AIMD’s scalability points restrict its use in biomolecular research. Machine studying power fields (MLFFs), educated on DFT-level knowledge, promise accuracy at decrease prices, although generalization throughout different molecular conformations stays difficult.
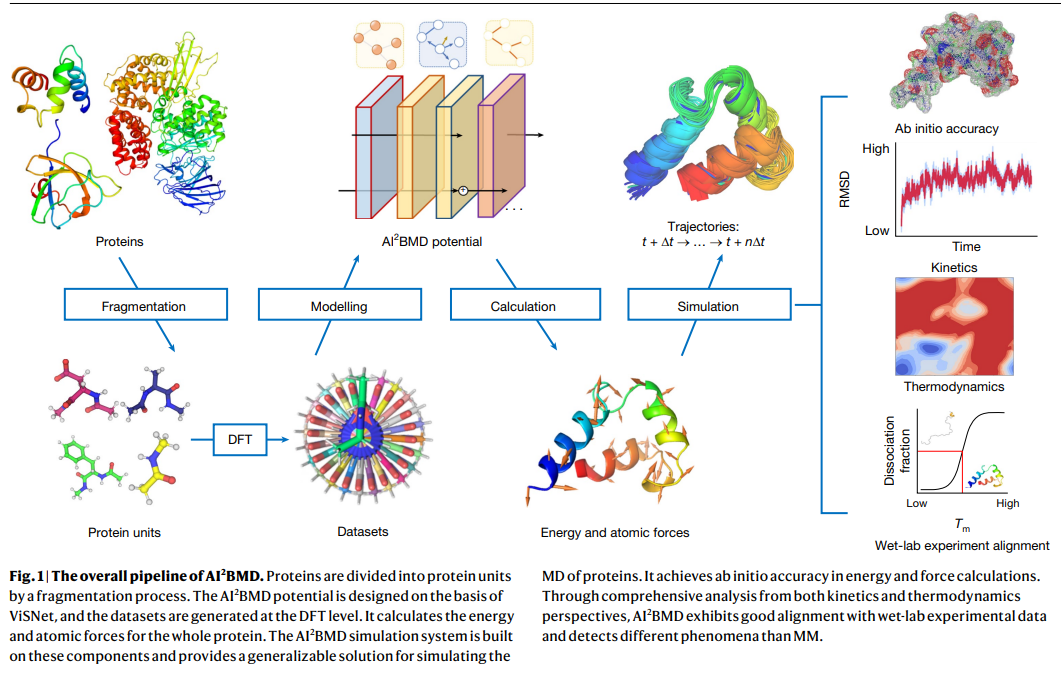
Researchers from Microsoft Analysis in Beijing launched AI2BMD, an AI-based system for simulating giant biomolecules with ab initio accuracy. AI2BMD makes use of a protein fragmentation method and a machine studying power discipline, permitting it to precisely compute vitality and forces for proteins with over 10,000 atoms. This technique is vastly extra environment friendly than conventional DFT, lowering simulation occasions by orders of magnitude. AI2BMD can conduct a whole bunch of nanoseconds of simulations, capturing protein folding, unfolding, and conformational dynamics. Its thermodynamic predictions align intently with experimental knowledge, making it a beneficial device for complementing moist lab experiments and advancing biomedical analysis.
The protein fragmentation method builds on the foundational construction of amino acids in proteins, the place every amino acid accommodates a primary chain of atoms (Cα, C, O, N, and H) and a definite aspect chain. To create a mannequin that applies broadly to numerous proteins, every amino acid is handled as a dipeptide, capped with Ace and Nme teams at its ends. This method, primarily based on overlapping fragments of dipeptides, helps guarantee complete protein protection. Utilizing a sliding window, protein chains are divided into these dipeptides, the place every fragment contains primary chain atoms and partial atoms from adjoining amino acids. This technique precisely calculates protein energies and atomic forces by including hydrogens as required for Cα bonds and optimizing positions utilizing a quasi-Newton algorithm. This generalizable technique permits the systematic software to all proteins, lowering complexities whereas maximizing mannequin accuracy.
The coaching dataset for the AI2BMD potential includes sampling tens of millions of dipeptide conformations to seize the variability in protein constructions. A deep studying mannequin referred to as ViSNet was educated utilizing this in depth dataset to foretell the vitality and atomic forces primarily based on atomic numbers and coordinates. The mannequin used particular hyperparameters to optimize accuracy and was educated with early-stopping strategies. Simulations primarily based on the AI2BMD potential are processed utilizing a cloud-compatible AI-driven simulation program, enabling versatile deployment throughout computing environments. This technique helps parallelized simulation processes and mechanically preserves progress on cloud storage, guaranteeing sturdy and environment friendly dealing with of protein dynamics modeling.
AI2BMD showcases important potential in protein property estimation, particularly for thermodynamic evaluation of fast-folding proteins. AI2BMD might categorize constructions into folded and unfolded states by simulating varied protein sorts and precisely predicting potential vitality values. Its melting temperature (Tm) estimations for proteins just like the WW area and NTL9 intently matched experimental knowledge, regularly outperforming conventional molecular mechanics (MM) strategies. Moreover, AI2BMD’s calculations totally free vitality (ΔG), enthalpy, and warmth capability had been extremely in keeping with experimental findings, reinforcing its accuracy. This robustness in thermodynamic estimation highlights AI2BMD’s worth as a complicated device for protein evaluation.
Along with thermodynamics, AI2BMD proved efficient in alchemical free-energy calculations, equivalent to pKa prediction, and is efficacious in biochemical analysis. In contrast to conventional QM-MM strategies that limit calculations to preset areas, AI2BMD’s ab initio method permits full-protein modeling with out boundary inconsistencies, making it versatile for advanced proteins and dynamic states. Though AI2BMD’s pace remains to be slower than classical MD, future optimizations and purposes to different biomolecular techniques might improve its effectivity. AI2BMD’s adaptability makes it a promising device for drug discovery, protein design, and enzyme engineering, providing extremely correct simulations for varied biomolecular purposes.
Try the Paper and Particulars. All credit score for this analysis goes to the researchers of this undertaking. Additionally, don’t overlook to comply with us on Twitter and be part of our Telegram Channel and LinkedIn Group. For those who like our work, you’ll love our e-newsletter.. Don’t Overlook to hitch our 55k+ ML SubReddit.
[Sponsorship Opportunity with us] Promote Your Analysis/Product/Webinar with 1Million+ Month-to-month Readers and 500k+ Group Members
Sana Hassan, a consulting intern at Marktechpost and dual-degree scholar at IIT Madras, is keen about making use of expertise and AI to deal with real-world challenges. With a eager curiosity in fixing sensible issues, he brings a contemporary perspective to the intersection of AI and real-life options.
[ad_2]
Source link


