

Conventional molecular representations, primarily centered on covalent bonds, have uncared for essential elements like delocalization and non-covalent interactions. Current machine studying fashions have utilized information-sparse representations, limiting their capacity to seize molecular complexity. Whereas computational chemistry has developed strong quantum-mechanical strategies, their utility in machine studying has been constrained by calculation challenges for complicated programs. Graph-based representations have offered some topological data however lack quantum-chemical priors.
The rising complexity of prediction duties has highlighted the necessity for higher-fidelity representations. This work addresses these gaps by introducing stereo electronics-infused molecular graphs (SIMGs), which incorporate quantum-chemical interactions. SIMGs intention to boost the interpretability and efficiency of machine studying fashions in molecular property predictions, overcoming the restrictions of earlier approaches and offering a extra complete understanding of molecular habits.
Molecular illustration is essential for understanding chemical reactions and designing new supplies. Conventional fashions use information-sparse representations, that are insufficient for complicated duties. This paper introduces stereoelectronics-infused molecular graphs (SIMGs), incorporating quantum-chemical data into molecular graphs. SIMGs improve conventional representations by including nodes for bond orbitals and lone pairs, addressing the neglect of important interactions like delocalization and non-covalent forces. This method goals to offer a extra complete understanding of molecular interactions, enhancing machine studying algorithms’ efficiency in predicting molecular properties and enabling analysis of beforehand intractable programs, similar to complete proteins.
The researchers employed Q-Chem 6.0.1 and NBO 7.0 for calculations utilizing a high-throughput workflow infrastructure. They performed Pure Bond Orbital evaluation to quantify localized electron data, excluding Rydberg orbitals. The workforce launched Stereo Electronics-Infused Molecular Graphs (SIMGs), incorporating stereoelectronic results and representing donor-acceptor interactions. Their mannequin structure stacked a number of graph neural community blocks with graph consideration layers and ReLU activation, addressing over-smoothing points in multi-layer networks. Efficiency analysis centered on lone pair classification and bond-related process predictions, demonstrating excessive accuracy and a 98% reconstruction fee of ground-truth prolonged graphs.
The mannequin demonstrated distinctive efficiency throughout numerous prediction duties, reaching excessive accuracy in classifying lone pair portions and kinds. It efficiently reconstructed the ground-truth prolonged graph in 98% of circumstances. Node-level duties confirmed outstanding efficiency, with atom-related predictions reaching wonderful R² scores and low MAEs and RMSEs. Lone pair predictions, particularly for s and p-character, achieved wonderful scores, whereas d-prediction duties confirmed barely decrease efficiency as a consequence of restricted knowledge.
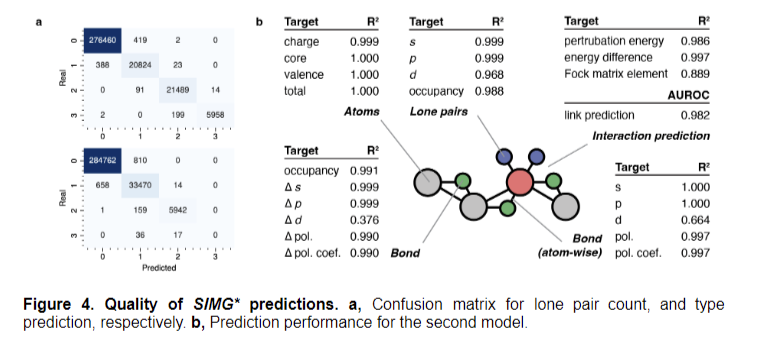
Bond-related process predictions had been favorable, significantly for hybridization characters and polarizations. Efficiency positively correlated with interplay pattern abundance. The F1 rating ensured unbiased measurements for imbalanced classifications, highlighting the mannequin’s effectiveness in capturing long-range interactions. These outcomes underscore the profitable integration of stereoelectronic results into molecular graphs, considerably enhancing the mannequin’s predictive capabilities throughout numerous molecular properties whereas additionally addressing challenges related to d-character predictions.
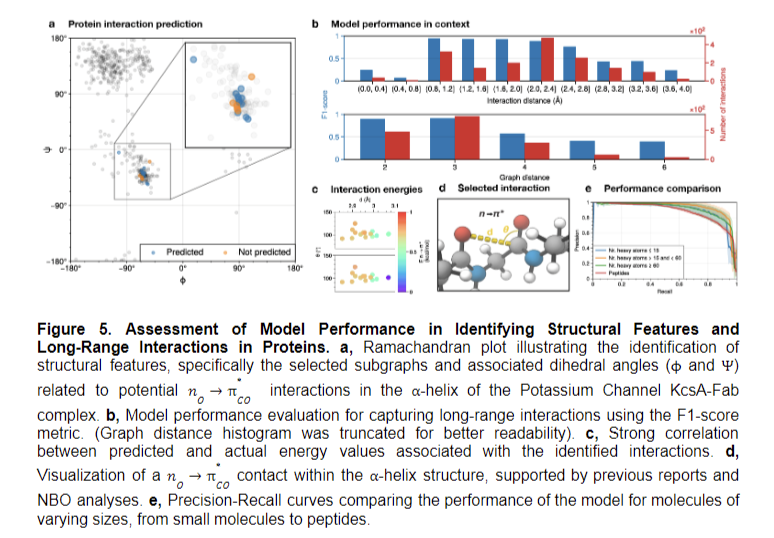
The examine concludes that incorporating stereoelectronic interactions into molecular graphs considerably enhances machine-learning mannequin efficiency, enabling an in depth understanding of molecular properties and behaviors. This method permits predictions for beforehand inaccessible molecules, together with complicated organic buildings. The brand new illustration facilitates high-throughput Pure Bond Orbital evaluation, probably accelerating theoretical chemistry analysis. The tailor-made double-graph neural community workflow allows the broad utility of discovered representations. These findings recommend additional exploration of stereoelectronic results may result in extra refined fashions, increasing purposes in drug discovery and supplies science. The examine demonstrates the potential for superior molecular representations to revolutionize predictive capabilities in chemistry and associated fields.
Try the Paper and GitHub. All credit score for this analysis goes to the researchers of this mission. Additionally, don’t neglect to observe us on Twitter and be a part of our Telegram Channel and LinkedIn Group. For those who like our work, you’ll love our publication..
Don’t Neglect to affix our 48k+ ML SubReddit
Discover Upcoming AI Webinars right here

Shoaib Nazir is a consulting intern at MarktechPost and has accomplished his M.Tech twin diploma from the Indian Institute of Know-how (IIT), Kharagpur. With a powerful ardour for Knowledge Science, he’s significantly within the numerous purposes of synthetic intelligence throughout numerous domains. Shoaib is pushed by a want to discover the newest technological developments and their sensible implications in on a regular basis life. His enthusiasm for innovation and real-world problem-solving fuels his steady studying and contribution to the sphere of AI
 Principles with Tool Integration Support")
")






{kind=link}